

Design of a New Topological Approach for the Prediction of Protein-Protein Interactions
Author: Leonardo Martini, Sapienza University of Rome
Protein-Protein Interactions (PPIs) play an essential role in several biological processes. In many cases, proteins perform essential functions by interacting to constitute protein complexes. Identifying new Protein-Protein interactions is thus crucial in understanding cells' biological mechanisms. Furthermore, the knowledge of the interactions is helpful for applications such as drug-repurposing (which leverages network topology to predict drug-disease associations) or for assessing disease-gene prioritisation (which leverages the PPI network to find new candidate disease genes).
Therefore, charting protein-protein interaction maps remains a fundamental goal in biological research.
Protein-protein interactions can be directly discovered by applying pull-down assay methods, the execution of yeast two-hybrid screens, or by purifying protein complexes tagged in vivo. Together with inherent advantages, these methods are all labour and time-consuming and have a high cost.
Computational tools capable of identifying prospective protein-protein interactions can be used to choose the protein-protein interactions to test with the resources demanding biochemical methods.
Computational methods based on the topology of the PPI network are inexpensive computational tools able to identify missing interactions inside an interactome.
The general problem of identifying new links in a network is known as the "link prediction" problem [2, 3]. Since its definition in 2007, several methods have been developed to address it; we refer the reader to some surveys [4–6].
Interestingly, link prediction methods based on paths of length two are particularly suitable in predicting missing links in social networks but, at the same time, are very inadequate in predicting interactions between proteins.
Barabasi et al. (2019) [1] show that this effect is related to the fact that the methods based on paths of length two are more suitable in scoring the similarity between proteins, in terms of the types of interactions they perform, than in scoring the likelihood of an interaction between proteins directly. On the contrary, methods based on the paths of length three are more suitable for scoring (directly) the likelihood of an interaction between proteins. Since Protein-Protein interactions often require complementary interfaces [7, 8] (i.e. complementary 3D structures), the authors showed that paths of length three capture this complementarity effect, identifying similar proteins/nodes to the known partners of a given node/protein.
Recently, the International Network Medicine Consortium (www.network-medicine.org) benchmarked the ability of new and old network-based methods to predict PPIs across different interactomes, including a synthetic interactome generated from the human interactome and the human interactome itself.
According to the computational validation performed on the human interactome, the best topological method resulted in a new method called "Maximum-Proteins-Similarity(Topological)": MPS(T).
MPS(T) is a topological three-length path method that scores the potential interaction between proteins by scoring the similarity (in terms of the types of interactions) using a well-known two-length path method, the topological Jaccard similarity between nodes.
More formally, given an interactome and two nodes 'u' an 'v' representing proteins inside the interactome, the topological Jaccard similarity 'J(u, v)' is defined as:
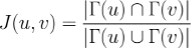
where 𝚪(u) is the set of u's neighbours in the interactome.
Considering the Jaccard similarity between two nodes as an indicator of the similarity of the interfaces of the two represented proteins, the MPS(T) method scores the complementarity of the proteins' binding sites and, consequently, the possibility of interaction according to the following formula:
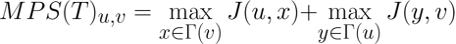
The following heat maps show the performances reached by each method tested in [9] through the computational validation performed on the human and synthetic interactomes.
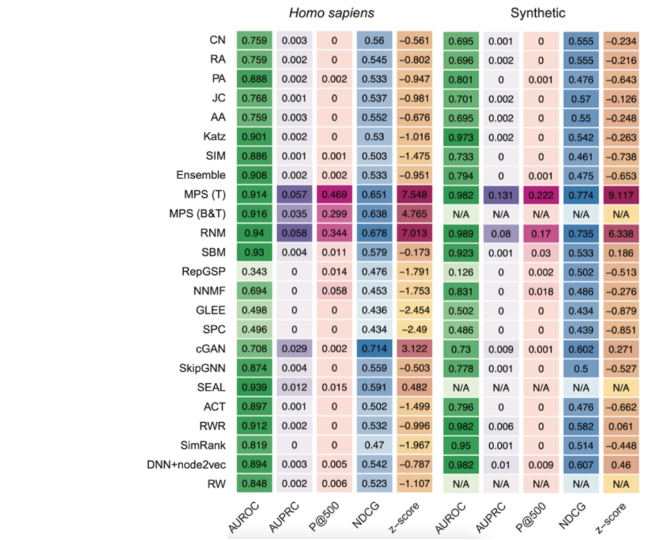
The MPS(T) method obtained the highest z-score in both the interactomes mainly thanks to its performance in the p@500 metric for the "Homo sapiens" interactome and the p@500, nDCG, and AUPRC metrics for the second network.
Moreover, from the top 500 PPIs predicted by MPS(T) from the "Homo sapiens" interactome, 358 were successfully tested by the yeast two-hybrid assay (Y2H), with 272 testings positive, yielding a precision of 75.97% [9].
Bibliography:
Kovács IA, Luck K, Spirohn K, Wang Y, Pollis C, Schlabach S, et al. Network-based prediction of protein interactions. Nature communications. 2019;10(1):1–8.
Liben-Nowell D, Kleinberg J. The Link Prediction Problem for Social Networks. In: Proceedings of the Twelfth International Conference on Information and Knowledge Management. CIKM'03. New York, NY, USA: Association for 441 Computing Machinery; 2003. p. 556–559. Available from: https://doi.org/10.1145/956863.956972
Liben-Nowell D, Kleinberg J. The link-prediction problem for social networks. Journal of the American Society for Information Science and Technology. 2007;58(7):1019–1031. doi:10.1002/asi.20591
Lu L, Zhou T. Link prediction in complex networks: A survey. Physica A: Statistical Mechanics and its Applications. 2011;390(6):1150–1170. doi: https://doi.org/10.1016/j.physa.2010.11.027
Martínez V, Berzal F, Cubero JC. A survey of link prediction in complex networks. ACM computing surveys (CSUR). 2016;49(4):1–33.
Kumar A, Singh SS, Singh K, Biswas B. Link prediction techniques, applications, and performance: A survey. Physica A: Statistical Mechanics and its Applications. 2020;553:124289. doi: https://doi.org/10.1016/j.physa.2020.124289
Keskin, O., Tuncbag, N. & Gursoy, A. Predicting protein-protein interactions from the molecular to the proteome level. Phys. Biol. 2, S1 (2005).
Szilágyi, A., Grimm, V., Arakaki, A. K. & Skolnick, J. Prediction of physical protein-protein interactions. Chem. Rev. 116, 4884–4909 (2016).
Wang XW, Madeddu L, Spirohn K, Martini L, Fazzone A, Becchetti L, et al. Assessment of community efforts to advance computational prediction of protein-protein interactions. bioRxiv. 2021. Doi: https://doi.org/10.1101/2021.09.22.461292